OPEN-ACCESS PEER-REVIEWED
REVIEW ARTICLE
Roland J.W. Meesters
MLM Medical Labs GmbH, 41066 Moenchengladbach, Germany.
Reviews in Separation Sciences. Vol.1. No.1. pages 34-46 (2019).
Published 15 October 2019. https://doi.org/10.17145/rss.19.004 | (ISSN 2589-1677).
*Correspondence:
Meesters RJ. . MLM Medical Labs GmbH, Dohrweg 61, 41066 Moenchengladbach, Germany.
Editor: Dr. Inas Abdallah, Faculty of Pharmacy, University of Sadat City, Egypt.
Open-access and Copyright:
©2019 Meesters RJ. This article is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Funding/Manuscript writing assistance:
The author has no financial support or funding to report and declares also that no writing assistance was utilized in the production of this article.
Competing interest:
The author has declared that no competing interest exist.
Article history:
Received 23 June 2019, Revised 10 August 2019, Accepted 14 August 2019.
Abstract
Volumetric absorptive microsampling (VAMS) is a novel microsampling technology that is frequently used for the collection of biofluids for the application in a plethora of bioanalytical applications and assays. Recently, few papers have reported the successful application of VAMS in untargeted as well in targeted metabolomics. VAMS in metabolomics can become in the near future a very promising microsampling technique, but to that moment, some application-related aspects of VAMS in the metabolomics workflow still needs some in depth investigation and evaluation. In this mini-review article, the application of VAMS in metabolomics, procedures applied and first results obtained are described, evaluated and discussed.
Keywords
Microsampling, Metabolomics, Volumetric Absorptive Microsampling, Blood, Mini-review.
1.0 Introduction
Metabolomics is the system level study of metabolism based on the identification and quantification of small-molecules (approx. <1500 Da) in biological systems [1, 2]. Metabolites play an important role in biological systems because they are the end-point of cellular processes as also building blocks of proteins, DNA, RNA and cell membranes [3]. Therefore, metabolomics is dedicated as the end-point of the “omics”-cascade that includes genomics, transcriptomics, proteomics and metabolomics [4]. Metabolite (concentrations) can offer important information on biochemical pathway functionality and they allow the interpretation of interactions of biological and environmental factors that contribute to the metabolomic phenotype of an organism [4]. Metabolomics can be conducted by two different methodological approaches. On the one hand, there is the global metabolomics approach (also called untargeted metabolomics) and on the other hand, there are targeted metabolomics. Targeted metabolomics aims to quantify a specific group or type of metabolites [2] that are involved in specific metabolic pathways or that have special functions [5].
In contrast to the targeted metabolomics approach is the aim of untargeted metabolomics the detection and identification of the complete set of metabolites in a biofluid, in a tissue or cells. As a result, thousands of peaks can be detected and identified by the use of metabolite libraries (i.e. Human Metabolome Database, METLIN, FiehnLib, and others) in biological samples [6]. Untargeted metabolomics has become increasingly applied in biomarkers discovery studies looking for new potential disease biomarkers.
In metabolomics (targeted and untargeted), a wide variety of biological sample types (matrices) can be employed. Blood and urine are the most common biofluids used and depending on the type of study and/or clinical research questions to be answered either one or even both biofluids are used. Blood samples will always provide a snapshot of the metabolic state and provide information related to biochemical catabolic and anabolic processes. Unlikely like blood, urine metabolomics will only give information on excreted polar metabolites related to biochemical catabolic processes.
Obtaining high-quality biological samples is crucial for metabolomics, and assuring this will require special attention. Sampling procedures for biological sample collection have to assure sample quality at any moment, especially in the collection of clinical samples regarding prevention of metabolite degradation or formation [7]. In many studies, blood is collected by venipuncture, although also other alternative less invasive blood collection techniques are used. The use of biofluid microsampling techniques has become lately widespread applied throughout the field of analytical and bioanalytical chemistry. The so-called dried blood spots microsampling technique (DBS) has already been applied for over 40 years on a large scale in the screening of inborn errors of metabolism of newborns [8, 9]. Next, to this application, DBS is very popular in the collection of blood samples for pharmaceutical analysis, drug development [10] and DBS were recently also with success applied in proteomics [11,12] and metabolomics [13].
In contrast to venipuncture, are microsampling techniques such as DBS relatively simple, safe and fast but there is one more valuable advantage. The most major advantage of DBS is that DBS potentially enables at-home blood sampling by patients while blood sampling by venipuncture is only possible by trained medical personnel in medical facilities. At-home sampling is often used for therapeutic drug monitoring and for patient’s adherence testing. However, despite all the advantages, DBS microsampling also presents quantitative bioanalysis with a number of issues and challenges, which on their hand potentially affect the quality of bioanalytical assays. Main issues of concern are the hematocrit effect (Ht-effect) and the DBS sample inhomogeneity, both issues will result in the introduction of an assay bias (positive or negative). Approximately five years ago a novel microsampling technique was introduced, the so-called volumetric absorptive microsampling (VAMS) [14]. In short time, VAMS became applied the analysis of small molecules in various biofluids [15, 16], protein analysis [17], ultra-trace analysis of metals [18] and environmental contaminants in blood [19]. VAMS was in principle developed to circumvent the problems DBS microsampling has with regard to the Ht-effect and inhomogeneity of the dried samples. VAMS technology is commercially marketed under the brand name MITRA® devices, which are an FDA class 1 CE/IVD device. A MITRA® device contains a porous tip for the accurate and reproducible collection of a fixed volume of blood through the principle of wicking [20]. MITRA® devices are available for the collection of different volumes of biofluid. At the moment, only a few papers report the application of VAMS for biofluid collection in metabolomics (untargeted and targeted studies).
In the following sections the VAMS technology, the challenges regarding the addition of an IS, solvent extraction of MITRA® devices, the effect of hematocrit on analyte recovery, analyte stability and quality assurance in metabolomics applying VAMS will be presented and discussed. Moreover, the present mini-review paper will provide the reader with a summary of recently reported results and will also present a short future perspective on the pros and cons of the application of VAMS in metabolomic studies.
2.0 Volumetric absorptive microsampling
Denniff and Spooner reported the first application of VAMS technology where it allowed an easy and fast way to collect a volumetric blood sample for quantitative assays [14]. The difference between VAMS and DBS microsampling is that with VAMS it is possible to accurately collect a precise volume of whole blood (CV 3.6%) [14] independent from the hematocrit, which is not possible with DBS. The MITRA® device consists out of two parts: i) a plastic sample handler, and ii) the sample absorbent tip. The absorbent tip is made from a special polymer which has hydrophilic properties. The absorbent tip contains pores that are specially designed to absorb a fixed volume of blood by capillary action (wicking). The MITRA® device can be purchased in different packing units such as a cartridge (2 devices), clamshell (2-4 devices) or a 96 device rack and three types of MITRA® devices are for the collection of 10, 20 and 30 µl of biofluid. For a correct and reproducible collection of a certain amount of blood, the device has to be held under a 45 degrees angle to the exit point of the blood by dipping the absorbent tip into the streaming blood. The absorbent tip directly absorbs blood and will be saturated with blood within a few seconds after the start of the blood collection. Important for the collection of a constant blood amount is that the absorbent tip should not be immersed into the blood past the tip shoulder because this will result in the collection of excess blood, mainly on the plastic handler. After the collection of the blood, the device absorbent tips need to be let too dry for at least two hours at ambient temperature. Thereafter the dried MITRA® devices can be directly used for analysis or transportation to a laboratory or when needed storage until further sample processing and analysis. For the extraction of the analyte(s) of interest the absorbent tip can be removed from the plastic handler or extracted while still attached to the plastic handler. Figure 1 presents the MITRA® device before collection- and after the absorption of the blood sample.
MITRA® devices can be used for biofluid collection by not medically trained individuals. This means that patients or care-takers in an at-home based environment can collect blood or other types of biofluids. A great example of a home-based application is blood sampling by patients using MITRA® devices for the monitoring of HbA1c in diabetic children [21,22].
3.0 Internal standard addition in VAMS
The incorporation of a surrogate or stable isotope labeled internal standard (IS) in assays using VAMS can be very challenging. Since VAMS are mainly applied in the collection of blood this section will have a focus on the use of MITRA® devices for blood collection. Nevertheless, equal challenges can be expected when other biofluids are sampled. Because the blood is absorbed by the absorbent tip through wicking from a freshly perforated skin, a reproducible addition of an IS before the blood is being absorbed by the sorbent tip is very challenging, so not from a practical point-of-view impossible. Because VAMS is a dried biofluid sampling technique the addition of an IS is not only a challenge for VAMS but also other microsampling sampling techniques such as DBS and capillary microsampling (CMS) using small glass capillaries.
Optimal analytical performance of an IS can only be guaranteed when the IS is added as early as possible into the sample preparation workflow. Ideally, before or directly after the collection of the biofluid. Only then, the internal can correct for analyte loss, degradation during collection and storage, and following sample preparation steps. Even when the addition of an IS would be practically possible at the moment of biofluid collection there would be a need for trained laboratory personnel. In an home-based patient setting the great advantage of VAMS regarding at-home self-sampling by patients will significantly be limited so not become impossible when the addition of an IS is for analysis is compulsory.
As a comparison, for the addition of an IS in VAMS a paper published previously on IS addition in DBS is discussed. Meesters et al. [23] studied the four ways of IS addition in DBS sample collection and preparation. For that purpose, the following moments for the addition of an IS in DBS preparation and extraction were evaluated by using a stable isotope labeled IS: i) the IS was mixed in an additional tube with blood and from the tube the DBS samples were prepared by pipetting the spiked blood sample, DBS was prepared, punched and extracted; ii) the DBS filter paper was impregnated with the IS by pipetting before the blood sample was added, punched and extracted; iii) the IS was pipetted on top of a DBS sample before punching and extraction, and iv) the IS was dissolved in the extraction solvent and punches from DBS samples were extracted. The results from the first experiments from experimental design demonstrated as well confirmed the expectation that the most reliable results for the analyte (recovery 107.9 ± 4.5%) were to be obtained when the IS was mixed with collected blood before the preparation of the DBS samples on the filter paper. No doubt exist that this experiment presents the “Gold Standard” principle of IS addition. Although the good results obtained, this experimental setup of IS addition would be very challenging in a real clinical study setup. Applying this setup would mean that the addition of IS has to occur at the moment the biofluid sample is collected what would be impossible or very challenging at that time-point for the majority of the clinical studies. This setup would without any doubt have the need of trained laboratory personnel at every sampling site. This procedure could potentially increase clinical study costs and would also be very challenging or even impossible in a home-based patient setting.
This would mean that one has to consider a different type of procedure for standard addition in VAMS. This could be a methodology that is based on a more practical and less challenging sample collection protocol. Knowing this, only the three other procedures for IS addition in VAMS are left, and one should select the procedure that is as well executable in the daily laboratory or clinical study practice and less challenging and therefore delivering reliable and reproducible results. The second most reliable solution for IS addition reported by Meesters et al. [23] was impregnation of the DBS filter paper with IS and addition of blood on the top of the dried IS. The analyte of interest could be recovered. This recovery rate is in principle acceptable for quantitative analysis (targeted metabolomics) in clinical studies for the majority of the analytes but the danger exists that when the recovery becomes lower or too low that this IS addition procedure will lead to discrimination of analytes with a concentration near the limit of detection (LOD) for untargeted metabolomics and near the limit of quantification (LLOQ) for targeted metabolomics because of its low recovery. The obtained relative “high recovery” of 87.5 ± 7.2 % was possible because the with IS impregnated filter paper was only covered by dried blood matrix from only one side of the DBS punch (side without dried blood). Because the side of the DBS sample where the dried blood was located was for the extraction solvent to extract the IS less available then the side of the filter paper where no dried blood was located. This effect is also expected to occur when the IS is absorbed on the absorbent tip of a MITRA® device but in this case, the extraction of the IS would be influenced by the dried blood located on top of the IS and it would lead to an overestimation of analyte concentrations. Nevertheless, the addition of an IS using this procedure could be suitable for VAMS, although it is at this moment not known what the impact of dried blood matrix covering the IS will be on the extraction of the IS and if there also exists a relationship between the IS recovery and the hematocrit of the collected blood sample. Furthermore, using VAMS for untargeted metabolomics, it would be impossible to determine this influence of the dried biofluid matrix on the analyte recovery. This is because in contrast to targeted metabolomics, in untargeted metabolomics the analysis and identification of a complete set of metabolites with different physicochemical properties is the aim. In this setting, it is not possible to determine the recovery rate for each metabolite when no information on which and the number of metabolites to expect is available.
Recently Kok et al. [24] reported the feasibility of IS (stable isotope labeled) addition by addition of the IS on the VAMS tip before the blood sample collection. Authors found that the addition of the IS did not significantly influence the maximum collected amount of blood nor did it influence the extraction of 36 compounds (amino acids and organic acids) in a targeted metabolomic study. Reported recoveries of the majority of the analyzed amino acids and organic acids ranged between 60 and 92 %, except for the following analytes the recoveries were significantly below 60 %; arginine 47%, 2-oxo-glutaric acid 5%, glyoxylic acid 23% and malic acid 23%. Authors concluded that VAMS could present great possibilities in metabolomics studies using multiple sample collections but that a targeted metabolomics study on mice has to be conducted to confirm the reported conclusions. Reported recoveries were obtained from blood collected from one healthy volunteer using a finger prick. It is not clear from the paper from Kok et al. [24] at which hematocrit value the recovery results for amino acids and organic acids were obtained. In conclusion, impregnation of the absorbent tip with the IS before blood collected provided satisfactory recovery rates but it is unclear if equal results would have been obtained if recoveries for analytes were determined at different hematocrit values.
A different study applying untargeted metabolomics conducted by Volani et al. [25] reported the addition of stable isotope labeled IS to the samples after extraction of the VAMS absorbent tips with solvent and centrifugation and filtration of the extract followed by evaporation of the solvent to dryness under vacuum at 35 ºC for 120 min in a vacuum evaporator. The study’s main focus was on the influence of solvent composition as well as the pH of the solvent used for the extraction of the VAMS absorbent tips. The samples were thereafter reconstituted in an acetonitrile/water mixture (50:50) containing stable isotope labeled amino acids. Authors evaluated different extraction protocols for the determination of the best pre-analytical conditions for VAMS-based metabolomics. Specific polar metabolites were studied and paper reports that a mixture of 70:30 acetonitrile/water yielded the best results based on the prerequisites selected by the authors of the paper.
In conclusion, it can be said that more research regarding the addition of ISs in VAMS will be needed to be able to develop a consensus on how and at what time point during the metabolomic workflow, the addition of an IS in VAMS is needed and applicable in metabolomics.
4.0 Metabolite extraction from absorbent tips
Extraction of metabolites from the VAMS absorbent tip can be easily achieved by detaching the absorbent tip from the holder or by immersing the tip when still mounted onto the tip-holder into an appropriate solvent or solvent mixture. Caution needs to be taken when the absorbent tip is removed from the tip-holder because removal of the absorbent tip could be a potential source of sample contamination. Extraction of analytes from the absorbent tip is a complex process where interactions between analyte-absorbance material, analyte-dried blood matrix need to be overcome by the extraction strength of the used solvent. The extraction process can be divided into two separate steps: i) the desorption of the analyte and ii) transport of the desorbed analyte by the solvent. The first step is believed to impact the recovery bias for the analyte while the second step in the extraction process is impacting the matrix related extraction bias [26]. It is obvious that when determining the best solvent for desorption/extraction of analytes from the absorbent tip, a trial-error protocol applying different solvents or solvent-solvent combinations is reported in published papers [26]. Mainly, hydrophilic solvents or combinations of hydrophilic solvents (methanol and acetonitrile) with or without water addition are used for obtaining high efficiency regarding desorption/extraction of analytes, leading to satisfactory analyte recovery rates. Cala and Meesters [27] applied only methanol for the extraction of VAMS absorbent tips without evaluation of the best extraction solvent or solvent mixture. In this study, they compared different microsampling techniques in a metabolic fingerprinting study and decided to use one single solvent to have a fair comparison of all microsampling techniques. In this study, the performances of two microsampling techniques (dried matrix on paper disks (DMPD) [10] and VAMS) were compared with conventional blood sampling. The comparison was based on the total number of metabolites detected by the different microsampling techniques using liquid blood as reference biofluid. The number of total identified metabolites applying the microsampling techniques were not significantly different from the number of identified metabolites in protein precipitated blood. The study is to the moment’s knowledge of the writing of this article the only reported study applying GC-MS detection of metabolites after two-step derivatization in combination with biofluid sampling applying VAMS (and DMPD). Compared to protein precipitated blood, were in DMPD and VAMS, 94 % and 93 %, respectively the total number of metabolites detected in protein precipitated blood to be detected. The untargeted metabolomics study conducted by Cala and Meesters could confirm the results published by Kong et al. [28] regarding the number of identified metabolites using conventional blood protein precipitation as well as microsampling techniques.
A different paper on the application of VAMS in metabolomics was published by Volani et al. [25]. The authors describe the application of sequential extraction of metabolites using different acetonitrile/water mixtures (70:30) at acidic (pH 2), neutral (pH 7) and basic pH (pH 9). The most promising results were obtained from the extraction of the absorbent tips with a neutral acetonitrile/water mixture because they observed that with a change of the pH of the extraction solvent from a neutral pH to an acidic or basic range the total number of identified metabolites, as well as the metabolic profile, detected decrease significantly. In this study basic extraction conditions altered the metabolomic profile with metabolites originating from erythrocytes, but also carboxylic acids could be better extracted, except for the organic acid citric acid which was better extracted at neutral pH, this because citric acid is a weak acid. With sequential extractions up to three consecutive steps, it was observed that approx. 80% of all extractable metabolites were desorbed/extracted in the first extraction step with ACN-H2O at pH 7. In the second extraction step, 17% of the total extractable metabolites and in the last third extraction the remaining 3% of the total extractable polar metabolites were extracted. The obtained extraction pattern was observed for the metabolites; amino acids, carboxylic acids, phosphorylated metabolites, carnitines, and other metabolites. The only exception to the 80-17-3% distribution of metabolites over the three extractions was detected for lipids. In the first extraction only approx. 50 % of the extractable lipids were detected while in the second and third extraction, 35% and 15%, respectively were detected. In general, multiple extraction steps of absorbent tips devices will lead to a better extraction recovery for all metabolites, leading to better coverage of the sample metabolome, this especially counts for untargeted metabolomics. Volani et al. [25] reported that a loss of 20% of polar metabolites by applying just a one-step extraction was acceptable, this because the applied extraction protocol was much faster and simpler to execute. One can image what the result of an untargeted metabolomics study will be when a potential (new) biomarker is present in the 20 % fraction or when the concentration of a (new) potential biomarker is near the LOD of the using bioanalytical method. Without any doubt, one should aim for the highest recovery of metabolites as possible to avoid the discrimination of metabolites that could present potential (new) biomarkers. Despite the good results obtained in this study, it is not clear at which hematocrit value these experiments were executed, more research regarding the influence of hematocrit and the impact on sequential extraction especially on the impact of the extraction methodology on the hematocrit based bias of VAMS tips is needed.
In conclusion, the application of different solvents or solvent mixtures at eventually different pH values will have a significant impact on the information that can be obtained from a metabolomics study as reported and confirmed by Volani et al. [25]. The impact of solvent selection on the total number of metabolites detected will depend on the type of solvent (solvent mixture) used as well as on the pH of the solvent or solvent mixture.
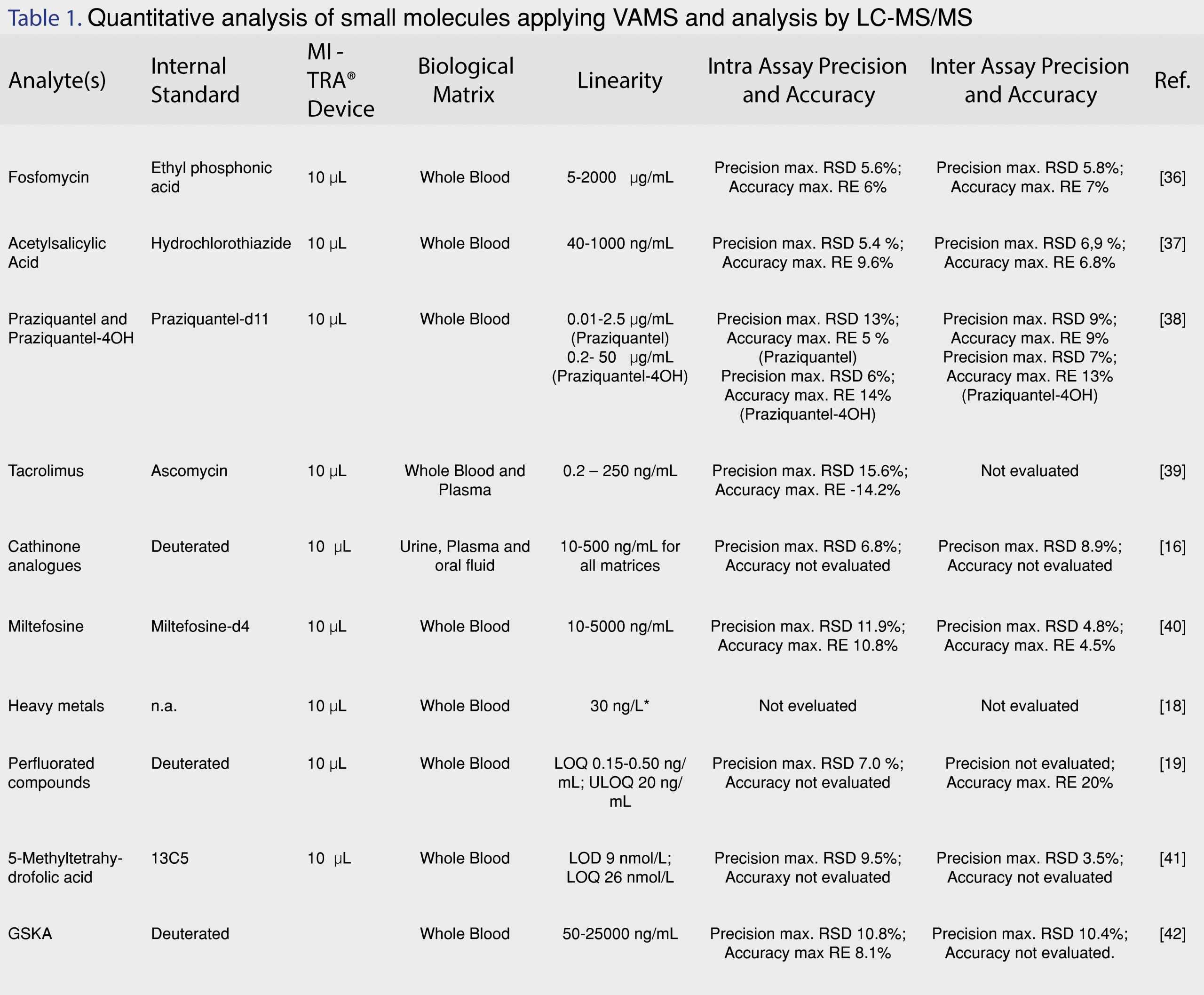
4.1 VAMS applications in quantitative analysis of small molecules
The application of the VAMS technology in quantitative analysis of small molecules in bioanalytical assays using mainly mass spectrometry for detection is demonstrated in Table 1. The information given in Table 1 does not provide a comprehensive overview of all publish VAMS applications but the table will provide a brief overview of some of the VAMS applications reported in recent literature. A more elaborate review on procedures and performances of VAMS methods was recently published by Protti et al. [26].
5.0 Effect of hematocrit
The extraction yield of metabolites from a VAMS absorbent tip is in comparison with DBS independent of the sample’s hematocrit. In DBS a positive bias is observed for high hematocrit values while a negative bias with low hematocrit values is observed [23]. VAMS is known for its hematocrit-independent absorption of a fixed volume of blood, providing a hematocrit-independent assay accuracy. In fact, some papers report an inverse correlation between the assay’s accuracy and hematocrit values. Applying VAMS, even with extreme hematocrit values (20 % and 70 %) no hematocrit dependent extraction efficiency is observed. Although this is not completely through, when extraction yields are not very high (ranging between 70 % to 75 %) the hematocrit can also in VAMS become an important factor. A severe hematocrit effect with VAMS was observed (approx. 60% bias) when hematocrit changed from 20 % to 70% [29]. The effect observed has been attributed to erythrocytes clogging the polymer pores and making the extraction of analytes more difficult. If so, the hematocrit will impact the assay’s accuracy and precision as hematocrit also does in DBS. The difference in hematocrit effect observed with VAMS and DBS is that with VAMS the hematocrit effect is a bias based on extraction efficiency while in DBS the hematocrit effect is a bias based on sample amount used for analysis.
Because VAMS is a perfect instrument for home-based patient sample collection there would be a need for the determination of the patient’s blood hematocrit since hematocrit can have a significant impact on the accuracy and precision of the analysis to a certain extent, especially in targeted metabolomics where the evaluation of data is made based on quantitative analysis.
6.0 Analyte stability
One of the great advantages of dried biofluid samples is the increased sample stability when compared with frozen biofluids. As has been reported in many papers, this advantage reduces significant costs of low-temperature storage and transportation [30]. Moreover, it is because of the increased sample stability of dried biofluid samples (DBS and VAMS) that this microsampling technique is perfect for a home-based patient sampling of blood or other biofluids. In metabolomics, it is very important that the samples used are stable in time during storage because degradation or formation of metabolites can significantly change the metabolomic profile, leading to wrong interpretation or results and conclusions regarding potential (new) biomarkers. Drying the MITRA® devices immediately after collection will increase to a certain extent the stability of especially unstable metabolites and will prevent the formation of degradation products of metabolites. In general, MITRA devices are dried for at least two hours after collection [15]. Kok et al. [24] studied the stability of different amino acids and organic acids in absorbent tips when stored under different storage conditions. Short-term storage stability was tested by storage of VAMS absorbent tips between 0.5 and 26 hours at room temperature. To have a reference, wet blood was stored at room temperature it was observed that an increase in the peak areas of the amino acids ornithine and arginine and the organic acids fumaric acid and lactic acid could be observed. These changes were not observed when dried VAMS absorbent tips were analyzed. With this simple experiment, it was proven the strength of this novel microsampling technology for application in metabolomics.
Next to short-term stability of metabolites also, long-term storage of amino acids and organic acids in VAMS absorbent tips for up to 15 days at room temperature were evaluated and results obtained were very satisfactory. Many of the evaluated amino acids appeared to be stable under these conditions except for the amino acids methionine and tyrosine. These amino acids were relatively unstable and demonstrated a decrease in the peak area of 19.2 % and 15.4%, respectively. In contrary to the average relatively high stability of amino acids in VAMS absorbent tips were organic acids reported as being less stable under selected storage conditions. For the organic acid malic acid, the tripeptide glutathione and the organic acids succinic acid and uric acid only a 4 day-stability could be guaranteed, whereas other metabolites were stable for minimal 7 days.
In general, can the stability of many metabolites be improved by storage of VAMS at lowered temperatures than room temperature, for example at -20, -30 or even at -80ºC. The storage of MITRA® devices at lowered temperatures will increase the stability of the analytes in VAMS until they are extracted by the solvent.
Volani et al. [25] evaluated the stability of the blood metabolome when using VAMS. It was reported that the changes in the metabolome were significantly when the MITRA® devices (n=30) were stored at room temperature. Storage of the devices (n=12) at a temperature of -80 ºC demonstrated that the metabolome was stable until a storage time of 6 months.
Even during the drying of the MITRA® devices, a significant change of the metabolome can occur. Some metabolites were rapidly degraded or generated during the first 48 h at room temperature, metabolites such as methionine sulfoxide, glutamic acid, and histidine but also other metabolites. In contrast, metabolites were also reported that increased at room temperature with the time of storage. Nevertheless, in metabolomics, it is a challenge to find a solution between stabilization of the metabolome of the collected samples and storage temperature, mostly low temperature (-20 or -80ºC) is the most applied approach [25]. The advantage of VAMS here is that for each sample multiple MITRA® devices can be stored and that the evaluation of freeze-thaw stability for frozen wet blood samples is not needed. Moreover, multiple freeze-thaw cycles might have a significant impact on analyte stability, which is eliminated by the extract-only-once possibility of MITRA®devices. Determination of metabolite stability regarding “freeze-thaw”-cycles is for MITRA® devices not possible because after extraction of the absorbent tip the MITRA® device has to be discharged.
Figures and Tables


[Click to enlarge]
7.0 Quality assurance in metabolomics using VAMS
The most commonly applied guidelines in the validation of bioanalytical methods are the guidelines published by the United States Food and Drug Administration (USFDA) titled “ Guidance for Industry: Bioanalytical Method Validation” [31]. These guidelines were developed for targeted drug analysis but the bioanalytical validation principles described in these guidelines can be adapted with care and some changes, to multi-compound targeted assays. Whilst these guidelines provide a good practical foundation for metabolomics system suitability and QC processes, one has to keep in mind that these guidelines were not designed for metabolomics and as such there are at this moment no harmonized guidelines for metabolomics available.
The study by Volani et al. [25] applied pooled QC samples prepared by pooling 30 µL of each extracted VAMS absorbent tip. The QC samples were used for the evaluation of intra-batch and inter-batch variability. A maximum RSD of 30% (according to FDA guidance) as acceptance criteria were used for this untargeted metabolomic study. LC-MS analysis was used and system suitability was studied by injection of the pooled QC samples (n=15) before analysis of the real samples. At the end of each sample sequence, blank samples were injected for testing carry-over. Unfortunately, the paper by Volani et al. doesn’t report any data on the performance of the pooled QC samples applied in the evaluation of LC-MS system suitability and intra-and inter-batch variability.
The study by Cala and Meesters [27] using GC-MS analysis in a comparative study between two different microsampling techniques and conventional protein precipitated blood samples applied pooled QC samples. QC samples were analyzed at the beginning of each batch and after the analysis of a set of five patient samples. This study applied real clinical samples originating from a healthy control group and a group of women diagnosed with breast cancer (between stage I and III). The reproducibility of the VAMS-QC samples was higher than for the DMPD-QC samples when the protein precipitated QC samples were used as a reference in terms of QC reproducibility. VAMS demonstrated a higher reproducibility then DMPD in all cases, particularly when CV was <10 %. Another important tool used by Cala and Meesters [27] for quality and reproducibility of the metabolomics data was the grouping of the QC samples in unsupervised models. PCA models were determined with metabolites having a CV of < 30 % in QC samples for the three blood microsampling techniques studied. The clustering of the QC samples was for all three different microsampling techniques very good and spontaneous separation of the control group and breast cancer group by biological variations demonstrated that all microsampling techniques performed well. A tighter grouping of the QC for the protein precipitated samples were observed demonstrating a higher reproducibility for this sample type. Cala and Meesters [27] reported that after the confirmation of the data quality by using QC reproducibility and grouping of QC samples in a principal component analysis OPLS-DA analysis was applied to elucidate the discrimination between the control group and breast cancer group using all three microsampling techniques. A good separation of both groups was observed and high model variables (R2 and Q2) were obtained for all evaluated microsampling techniques. OPLS-DA models were validated and cross-validated to estimate the predictive ability, and samples were predicted for 97% in protein precipitated samples and 96% and 96 % for DMPD and VAMS, respectively. It was suggested that both microsampling techniques DMPD and VAMS will yield similar results when applying protein precipitated blood. Finally, univariate analysis was applied to calculate p-values for each detected metabolite in the statistical comparison of the three sampling techniques. Cala and Meesters [27] reported that for precipitated blood, DMPD and VAMS there were 11,12 and 9 metabolites statistically different between the microsampling techniques, respectively. This study confirms the applicability of VAMS in untargeted metabolomics studies also when real clinical samples from a control group and a group of diseased subjects, in this case, breast cancer, were used to study the applicability. The study by Cala and Meesters [27] also confirms that microsampling of blood in clinical studies has great potential and will lead to a lower burden for healthy controls and patients included in clinical studies regarding the collection of blood or other biofluids. This burden will decrease due to a more simple and faster collection of blood samples but also lead to more pediatric studies where is a special need for more clinical research regarding certain diseases and were at this moment relative blood volume samples are collected which are in certain clinical settings not possible and hamper clinical studies and the development of new therapies and/or pharmaceutical drugs.
8.0 Future outlook
This mini-review presents an overview of the application of volumetric absorptive microsampling in metabolomics. Metabolomics has a huge potential in clinical and biomedical research fields such as disease metabolism studies [32], monitoring disease prognosis, diagnosis and therapy efficacy [33, 34], drug discovery [1, 35] and specialized metabolomics areas such as pharmacometabolomics. Microsampling techniques have gained growing interest for the collection of biofluids, this due to the small volume of biofluid employed (10 to 100 µL) this in contrast to conventional biofluid sampling where mostly several milliliters of blood are collected. Microsampling techniques are very useful for biofluid collection in special patient groups with patients having difficult-to-penetrate veins such as newborns, infants, the elderly and obese patients.
Volumetric absorptive microsampling (VAMS) is a microsampling technique that has the potential to become a very powerful low volume blood collection technique especially for special patient groups but also in regular patient groups. Volumetric absorptive microsampling has proven in a limited number of studies to be a great alternative tool in the collection of low volume blood samples. The use of VAMS for collection of blood samples is simple and fast and presents a possibility for inclusion of this microsampling technique of patient’s home-based blood collection. Compared with dried blood spots, VAMS presents several advantages such as hematocrit independent volumetric sampling of blood although a totally negligible impact of the hematocrit on the analysis of metabolites doesn’t exist.
Protti et al. [26] report in their review article some advantages beside the hematocrit independent collected sample volume of VAMS over DBS microsampling in bioanalysis. The easiness of workflow development and optimization is one of the strong points of VAMS compared with DBS. Furthermore, when correctly developed will a VAMS procedure be hematocrit indepent and will therefore demonstrate no hematocrit depent bias. Automation is straightforward when using VAMS compared to DBS where automation is more complicated but the main advantage of DBS over VAMS will always be the much lower price of DBS.
The application of VAMS in metabolomics has been only reported in a limited number of papers (Table 2). Although a limited number of papers describe the application of this novel microsampling technique the results reported by these papers are promising. One of the major drawbacks of dried blood spots (DBS), another microsampling technique, is the hematocrit effect and inhomogeneity of DBS samples when sampling whole blood samples. Introduction of VAMS has eliminated the hematocrit effect to a certain extent. A hematocrit influence has been also observed with VAMS but this effect exhibit only a significant impact when analyte extraction recoveries are relatively low (70-75%). This effect has been attributed to erythrocytes clogging up the pores of the porous polymer of the VAMS tip.
When applying VAMS in targeted metabolomics, one should be aware that that the recovey of metabolites might be depending on the haematocrit value of the blood sample. In order to obtain realiable biomarker concentration data, optimization of the extraction method of the analytes of interest is very important. Optimized extraction methods will overcome or limit to a certain the haematocrit impact on the accuracy of the biomarker concentration determination. Further improvement of the method’s accuracy can be reached by the usage and preparation of matrix matched calibrators and QC samples at a haematocrit value equal to or at the average range of the haematocrit of the expected clinical samples. In untargeted metabolomics of course optimization of extraction methods is complicated because optimization of the extraction recovery for a certain class of metabolites (lipids, sugars, amino acids etc.) might lead to the discrimination of other class(es) of metabolites. One could consider developing optimized extraction methods for certain or multiple classes of metabolites. The application of metabolite-class dependent extraction methods would on the one hand complicate the metabolomic workflow as well make the metabolomic analysis of the sample more expensive. On the other hand though, when using metabolite-class dependent extraction methods will to my point-of-view lead to an improved precision and more reliable data both resulting into the improvement of data quality for untargeted metabolomics studies.
In conclusion, VAMS clearly is a promise as a new microsampling technique when applied for sample collection in metabolomics. More metabolomics studies applying VAMS have to be conducted to really demonstrate the feasibility of this novel microsampling technique for metabolomics. Furthermore, the application of ISs has to be studied also especially in a real clinical study with real patients samples. The first results obtained by the addition of ISs prior to collection of blood was also very promising but was only evaluated for a limited group of metabolites, amino acids, and organic acids.
9.0 References
1. Mastrangelo A, Armitage EG, Garcia A, Barbas C. Metabolomics as a tool for drug discovery and personalised medicine. A review. Current topics in medicinal chemistry 14(23), 2627-2636 (2014). [CrossRef]
2. Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, Mclean JA. Untargeted Metabolomics Strategies-Challenges and Emerging Directions. Journal of the American Society for Mass Spectrometry 27(12), 1897-1905 (2016). [CrossRef]
3. Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nature reviews. Molecular cell biology 17(7), 451-459 (2016). [CrossRef]
4. Dettmer K, Hammock BD. Metabolomics–a new exciting field within the “omics” sciences. Environmental health perspectives 112(7), A396-397 (2004). [CrossRef]
5. Griffiths WJ, Koal T, Wang Y, Kohl M, Enot DP, Deigner HP. Targeted metabolomics for biomarker discovery. Angew Chem Int Ed Engl 49(32), 5426-5445 (2010). [CrossRef]
6. Patti GJ, Yanes O, Siuzdak G. Innovation: Metabolomics: the apogee of the omics trilogy. Nature reviews. Molecular cell biology 13(4), 263-269 (2012). [CrossRef]
7. Yin P, Lehmann R, Xu G. Effects of pre-analytical processes on blood samples used in metabolomics studies. Analytical and bioanalytical chemistry 407(17), 4879-4892 (2015). [CrossRef]
8. Denes J, Szabo E, Robinette SL et al. Metabonomics of newborn screening dried blood spot samples: a novel approach in the screening and diagnostics of inborn errors of metabolism. Analytical chemistry 84(22), 10113-10120 (2012). [CrossRef]
9. Guthrie R, Susi A. A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics 32 338-343 (1963).
10. Sandoval Parra MA, Rincon Pabon JP, Meesters RJW. Bioanalytical evaluation of dried plasma spot microsampling methodologies in pharmacokinetic studies applying Acetaminophen as model drug. Journal of Applied Bioanalysis 2(1), 25-37 (2016). [CrossRef]
11. Chambers AG, Percy AJ, Yang J, Camenzind AG, Borchers CH. Multiplexed Quantitation of Endogenous Proteins in Dried Blood Spots by Multiple Reaction Monitoring Mass Spectrometry. Mol Cell Proteomics doi:10.1074/mcp.M112.022442 (2012). [CrossRef]
12. Edwards RL, Griffiths P, Bunch J, Cooper HJ. Top-down proteomics and direct surface sampling of neonatal dried blood spots: diagnosis of unknown hemoglobin variants. Journal of the American Society for Mass Spectrometry 23(11), 1921-1930 (2012). [CrossRef]
13. Wilson I. Global metabolic profiling (metabonomics/metabolomics) using dried blood spots: advantages and pitfalls. Bioanalysis 3(20), 2255-2257 (2011). [CrossRef]
14. Denniff P, Spooner N. Volumetric absorptive microsampling: a dried sample collection technique for quantitative bioanalysis. Analytical chemistry 86(16), 8489-8495 (2014). [CrossRef]
15. Kok MGM, Fillet M. Volumetric absorptive microsampling: Current advances and applications. Journal of pharmaceutical and biomedical analysis 147 288-296 (2018). [CrossRef]
16. Mercolini L, Protti M, Catapano MC, Rudge J, Sberna AE. LC-MS/MS and volumetric absorptive microsampling for quantitative bioanalysis of cathinone analogues in dried urine, plasma and oral fluid samples. Journal of pharmaceutical and biomedical analysis 123 186-194 (2016). [CrossRef]
17. Andersen IKL, Rosting C, Gjelstad A, Halvorsen TG. Volumetric absorptive MicroSampling vs. other blood sampling materials in LC-MS-based protein analysis – preliminary investigations. Journal of pharmaceutical and biomedical analysis 156 239-246 (2018). [CrossRef]
18. Bolea-Fernandez E, Phan K, Balcaen L, Resano M, Vanhaecke F. Determination of ultra-trace amounts of prosthesis-related metals in whole blood using volumetric absorptive micro-sampling and tandem ICP – Mass spectrometry. Analytica chimica acta 941 1-9 (2016). [CrossRef]
19. Koponen J, Rudge J, Kushon S, Kiviranta H. Novel volumetric adsorptive microsampling technique for determination of perfluorinated compounds in blood. Analytical biochemistry 545 49-53 (2018). [CrossRef]
20. https://www.neoteryx.com
21. Verougstraete N, Stove V, Stove C. Wet absorptive microsampling at home for HbA1c monitoring in diabetic children. Clinical chemistry and laboratory medicine : CCLM / FESCC 56(12), e291-e294 (2018). [CrossRef]
22. Verougstraete N, Lapauw B, Van Aken S, Delanghe J, Stove C, Stove V. Volumetric absorptive microsampling at home as an alternative tool for the monitoring of HbA1c in diabetes patients. Clinical chemistry and laboratory medicine : CCLM / FESCC 55(3), 462-469 (2017). [CrossRef]
23. Meesters R, Hooff G, Van Huizen N, Gruters R, Luider T. Impact of internal standard addition on dried blood spot analysis in bioanalytical method development. Bioanalysis 3(20), 2357-2364 (2011). [CrossRef]
24. Kok MGM, Nix C, Nys G, Fillet M. Targeted metabolomics of whole blood using volumetric absorptive microsampling. Talanta 197 49-58 (2019). [CrossRef]
25. Volani C, Caprioli G, Calderisi G et al. Pre-analytic evaluation of volumetric absorptive microsampling and integration in a mass spectrometry-based metabolomics workflow. Analytical and bioanalytical chemistry 409(26), 6263-6276 (2017). [CrossRef]
26. Protti M, Mandrioli R, Mercolini L. Tutorial: Volumetric absorptive microsampling (VAMS). Analytica chimica acta 1046 32-47 (2019). [CrossRef]
27. Cala MP, Meesters RJ. Comparative study on microsampling techniques in metabolic fingerprinting studies applying gas chromatography-MS analysis. Bioanalysis 9(17), 1329-1340 (2017). [CrossRef]
28. Kong ST, Lin HS, Ching J, Ho PC. Evaluation of dried blood spots as sample matrix for gas chromatography/mass spectrometry based metabolomic profiling. Analytical chemistry 83(11), 4314-4318 (2011). [CrossRef]
29. Mano Y, Kita K, Kusano K. Hematocrit-independent recovery is a key for bioanalysis using volumetric absorptive microsampling devices, Mitra. Bioanalysis 7(15), 1821-1829 (2015). [CrossRef]
30. Meesters RJ, Hooff GP. State-of-the-art dried blood spot analysis: an overview of recent advances and future trends. Bioanalysis 5(17), 2187-2208 (2013). [CrossRef]
31. Center for Drug Evaluation and Research (CDER) Guidance for Industry: Bioanalytical Method Validation. (2018).
32. Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer research 66(18), 8927-8930 (2006). [CrossRef]
33. Armitage EG, Southam AD. Monitoring cancer prognosis, diagnosis and treatment efficacy using metabolomics and lipidomics. Metabolomics : Official journal of the Metabolomic Society 12 146 (2016). [CrossRef]
34. Wishart DS, Mandal R, Stanislaus A, Ramirez-Gaona M. Cancer Metabolomics and the Human Metabolome Database. Metabolites 6(1), (2016). [CrossRef]
35. Cuperlovic-Culf M, Culf AS. Applied metabolomics in drug discovery. Expert opinion on drug discovery 11(8), 759-770 (2016). [CrossRef]
36. Parker SL, Roberts JA, Lipman J, Wallis SC. Quantitative bioanalytical validation of fosfomycin in human whole blood with volumetric absorptive microsampling. Bioanalysis 7(19), 2585-2595 (2015). [CrossRef]
37 Kim Y, Jeon J-Y, Han S-H, Jang K, Kim M-G. Quantitative analysis of acetylsalicylic acid in human blood using volumetric absorptive microsampling. Trans Clin Pharmacol 26(1), 32-38 (2018). [CrossRef]
38. Kovac J, Panic G, Neodo A, Meister I, Coulibaly JT, Schulz JD, Keiser J. Evaluation of a novel micro-sampling device, Mitra™, in comparison to dried blood spots, for analysis of praziquantel in Schistosoma haematobium-infected children in rural Côte d’Ivoire. J Pharm Biomed Anal 151, 339-346 (2018). [CrossRef]
39. Kita K, Noritake K, Mano Y. Application of a Volumetric Absorptive Microsampling Device to a Pharmacokinetic Study of Tacrolimus in rats: Comparison with Wet Blood and Plasma. Eur J Drug Metab Pharmacokinet 44(1), 91-102 (2019). [CrossRef]
40. Kip AE, Kiers KC, Rosing H, Schellens JH, Beijnen JH, Dorlo TP. Volumetric absorptive microsampling (VAMS) as an alternative to conventional dried blood spots in the quantification of miltefosine in dried blood samples. J Pharm Biomed Anal 135, 160-166 (2017). [CrossRef]
41. Kopp M, Rychlik M. Assessing Volumetric Absorptive Microsampling Coupled with Stable Isotope Dilution Assay and Liquid Chromatography-Tandem Mass Spectrometry as Potential Diagnostic Tool for Whole Blood 5-Methyltetrahydrofolic Acid. Front Nutr 4, 9 (2017). [CrossRef]
42. Fang K, Bowen CL, Kellie J, Karlinsey M, Evans C. Drug monitoring by volumetric absorptive microsampling: method development considerations to mitigate hematocrit effects. Bioanalysis 10(4), 241-255 (2018). [CrossRef]
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License

