OPEN-ACCESS PEER-REVIEWED
HOW TO,…ARTICLE ?
Moucun Yuan1, Omnia A. Ismaiel1,2*, William R. Mylott Jr1
1PPD Laboratories, Richmond, VA, USA: 2Department of Analytical Chemistry, Faculty of Pharmacy, Zagazig University, Egypt.
Reviews in Separation Sciences. Vol.1. No.1. pages 47-55 (2019).
Published 15 October 2019. https://doi.org/10.17145/rss.19.005 | (ISSN 2589-1677).
*Correspondence:
Ismaiel OA. . PPD Laboratories, Richmond, VA, USA.
Editor: Dr. Theo Noij, Charles River Laboratories, The Netherlands/Avans University of Applied Sciences, Breda, The Netherlands.
Open-access and Copyright:
©2019 Yuan M et al. This article is an open access article distributed under the terms of the Creative Commons Attribution License (CC-BY) which permits any use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Funding/Manuscript writing assistance:
The authors have no financial support or funding to report and declare also that no writing assistance was utilized in the production of this article.
Competing interest:
The authors have declared that no competing interest exist.
Article history:
Received 19 July 2019, Revised 29 August 2019, Accepted 02 September 2019.
Abstract
Bioanalysis of protein-based therapeutics have historically used ligand binding immunoaffinity (LBA) approaches. However, recently LBA has been combined with Liquid Chromatography-Mass Spectrometry, resulting in the term hybrid (LBA)-LC-MS/MS techniques. Hybrid LC-MS/MS has successfully been applied to the bioanalysis of biotherapeutics and biomarkers. The basic concept of hybrid LC-MS/MS can be defined as combining LBA for the selective isolation/enrichment of the target analyte from the biomatrix and LC-MS for selective detection of the isolated analyte. In this article, we present a brief overview of How To develop a hybrid LBA-LC-MS/MS method for biotherapeutics and biomarkers with an emphasis on the general workflow and procedures.
Keywords
Ligand Binding Immunoaffinity, Hybrid LBA-LC-MS/MS, Biotherapeutics, Biomarkers, Method Development.
1.0 Introduction
Since the foundation of ligand binding assay (LBA), which was established by the work of Yalow and Berson and the work of Landsteiner [1], LBA has been and continues to be used as a standard approach for bioanalysis of therapeutic proteins due to its excellent sensitivity and high throughput capabilities. However, as biotherapeutics have become increasingly popular so have the complexities of the molecular constructs, leading to significant challenges for traditional LBA techniques. Recently, Hybrid LBA-LC-MS/MS has been shown to be a promising and successful approach for biotherapeutics and biomarkers [2-6], providing alternative analytical tools to meet the needs of the ever-changing molecular constructs. Hutchens and Yip were the first to evaluate affinity capture techniques for purifying macromolecules followed by matrix-assisted laser desorption/ionization (MALDI) time-of-flight mass spectrometry in the early 1990s, referred to as “surface-enhanced affinity capture mass spectrometry” [7], followed by the work of Nelson et.al. using the term “Mass Spectrometric Immunoassay” [8]. This work helped to prove the applicability of the Antigen-Antibody immunocapture concept for identification and quantification of target antigens using mass spectrometry. In general, Hybrid LBA-LC-MS/MS assays have the potential to offer some advantages over traditional LBAs [4,6,9] such as:
Greater structural information and selectivity
- Quantitative results based on intrinsic physicochemical properties
- Can be developed without the need for specialized reagents
- Multiplex analysis for multiple analytes or protein domains
- Wide dynamic range (≥3 logs), minimizing dilutions
- Less sensitive to cross-reactivity and matrix-related interferences
- The resolving power of chromatography combined with the MS selectivity, allows structurally and chemically similar analytes to be separated and distinguished from each other.
- LC-MS/MS technology is common in many labs, with an increase in labs using hybrid LC-MS/MS approaches
This article will briefly introduce the principle of the hybrid LBA/LC-MS/MS, and will focus on the basic concept, assay workflow description, and extraction/enrichment procedures.
1.1 Definition of the Hybrid LC-MS/MS
Hybrid LC-MS/MS can be considered a combination of traditional LBA enrichment strategies, for the selective isolation of the biotherapeutic from the biologic matrix, and LC-MS/MS for the detection and quantitation of the analyte of interest. Traditional sample preparation (such as protein precipitation or solid phase extraction) or affinity capture enrichment strategies can be used for the separation of biotherapeutics and biomarkers from the biologic matrix [9]. In some cases, based on the assay requirements, a direct digestion approach, without an immunoaffinity step, can also be used.
Broadly speaking, traditional sample preparation, direct digestion and affinity capture approaches combined with LC-MS/MS often are considered to fall under the hybrid LC-MS/MS definition [9].
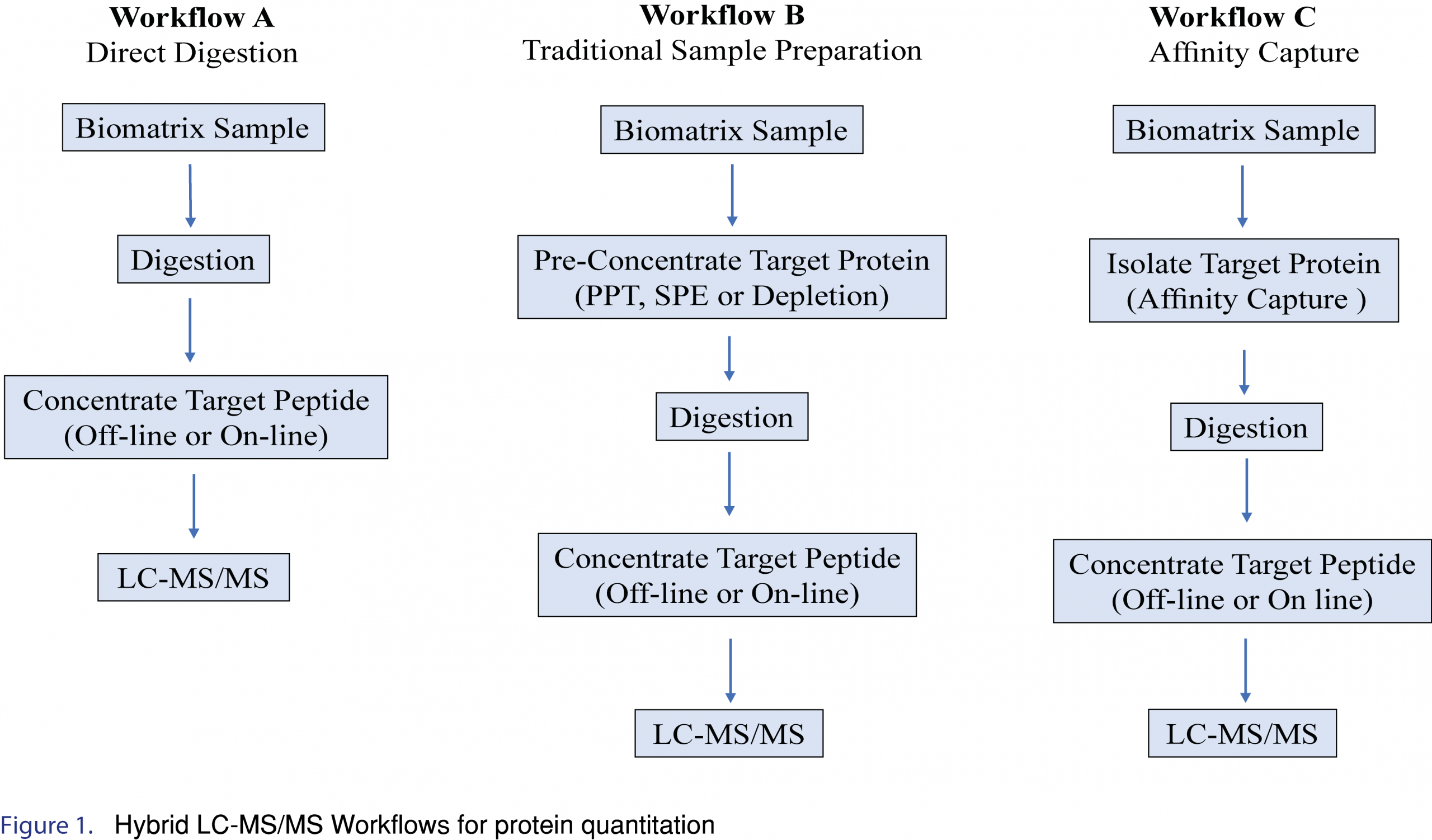
2.0 Hybrid LC-MS/MS Workflow
Examples of several Hybrid LC-MS/MS Workflows for protein quantitation are shown in Figure 1. In general, the simplest approach should be applied if the required selectivity/specificity, sensitivity, accuracy, and precision in the intended matrix can be achieved. However, direct digestion and traditional sample preparation approaches (such as protein precipitation or solid phase extraction) are limited in their ability to remove or reduce endogenous matrix components, often resulting in complicated or dirty extracts impacting assay sensitivity. In addition, these techniques often lose the ability to distinguish between different “forms” of the biotherapeutic protein, such as the ability to discriminate between free vs total, and often lose important structural information. Incorporating an immunoaffinity workflow offers many advantages, such as cleaner extracts, and maintaining structural information, and as such is the most useful/popular LC-MS/MS approach for large molecule isolation/detection. This article will focus on immunoaffinity-LC/MS/MS workflows.
3.0 Hybrid LC-MS/MS: Procedure and Materials
3.1 In silico analysis
Quantification of biotherapeutics by LC-MS/MS is primarily based on enzymatic digestion of enriched target protein followed by quantification of unique amino acid sequences (surrogate peptides). The surrogate peptides should be unique to the analyte and not found in any endogenous protein that might be present in the sample.
To facilitate the identification of candidate surrogate peptide sequences, the sequence of the target protein needs to be known. Some of the common protein sequences can be downloaded from online databases, such as the International Immunogenetics information system® website (http://www.imgt.org). In silico digestion using different proteases (such as trypsin) is applied on the protein amino acid sequence and the resulting candidate surrogate peptide sequences are searched (BLAST® Basic Local Alignment Search Tool) against the intended species and or other animal species protein sequences using an online database such as the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov) to identify surrogate peptides that are proteotypic. Often many proteotypic peptides are identified, resulting in the need to narrow the number of candidates to be evaluated. Typically, short peptides (fewer than 5 amino acids) and large peptides (>20 amino acids) are either excluded or given a low ranking in the candidate peptide pool. Peptides containing glycosylation consensus sequences or sequences that are susceptible to post-translational modifications, such as deamidation, oxidation or isomerization, should be avoided.
Once the initial in silico candidates have been identified, the list may need further refinement. The multiplexing nature of LCMS allows multiple candidate peptides to be evaluated simultaneously (e.g. from multi-domains for quantitative and/or confirmatory purposes). The candidate surrogate peptide chromatographic and mass spectrometric (MS) characteristics are evaluated. The peptides with favorable selectivity and sensitivity are considered for further evaluation/optimization. Surrogate peptides are typically classified as either 1). signature surrogate peptides, or 2). Universal surrogate peptides. A detailed description for each type will be described in the following sections.
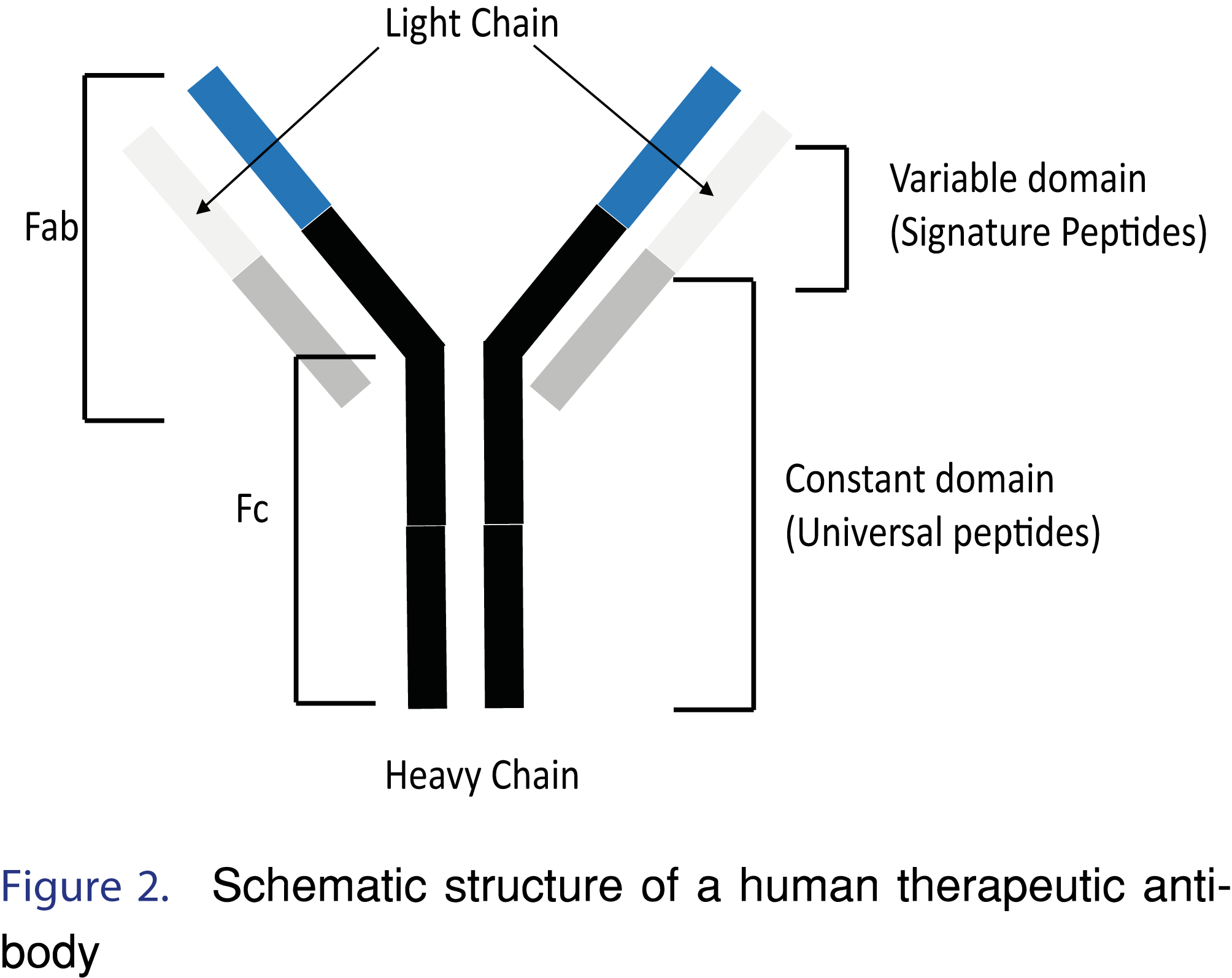
Signature surrogate peptides have amino acid sequences that are unique to the target biotherapeutic (e.g. peptide from the variable domain of therapeutic antibody) (Figure 2). Signature peptides provide excellent selectivity and are often the only choice when the intended matrix is human. However, since signature peptides are unique to a therapeutic, de novo bioanalytical development, starting with the in-silico analysis, is often required for new therapeutics [2]. For example, FTISADTSK and IYPTNGYTR are signature surrogate peptides from the complementarity determining region (CDR) of trastuzumab that have been used in quantification of trastuzumab in human serum [10].
Surrogate peptides from different regions (e.g. heavy chain, light chain, CDR, etc.) can also provide important structural information about the target protein. As an example, for the monoclonal antibody Ipilimumab [11], two characteristic or signature tryptic peptides, GLEWVTFISYDGNNK (GLEW) and LLIYGAFSR (LLIY) derived from the CDR, have been used in our laboratory as surrogates for Ipilimumab quantitation. The GLEW peptide is from the heavy chain and the LLIY is from the light chain. Incorporating an immunocapture step, such as Protein A or Protein G, and using signature peptides from both the heavy and light chains provides information about overall structure. The immunocapture involves binding the Fc region of the mAb, and quantitation using signature peptides from both heavy and light chain would allow an assessment to be made about the intact structure by comparing the concentrations obtained using the heavy chain peptide (often referred to as total antibody) to the concentrations obtained using the light chain peptide (often referred to as intact antibody). While light and heavy chain dissociation is not expected for Ipiliumumab, the payloads for many antibody drug conjugates involve inter-disulfide conjugation, which involves breaking the covalent linkage between the various chains. As such in vivo dissociation of the light and heavy chains, which would have a significant impact on the mAb-target binding efficiency and thus drug efficacy, would lead to different concentrations for the total and intact antibody concentration.
An alternative approach to using signature peptides is to use surrogate peptides that are considered “Universal peptides”. Universal surrogate peptides are common to different human protein-based therapeutics, such as mAbs, and not present in the protein sequences of the animal species that are commonly used in pre-clinical studies. For example, the tryptic peptide amino acid sequence VVSVLTVLHQDWLNGK, is present in human constant domain (the heavy chain of the Fc region for human IgG1, IgG3 and IgG4 isotypes), and the tryptic peptide TVAAPSVFIFPPSDEQLK is present in human constant domain (light chain), Figure 2 [2,12]. Both human tryptic peptides are proteotypic in animal matrix, thus leading to the term “Universal peptide”- universal across therapeutic and universal across animal species. For example, universal surrogate peptides have been applied to the quantification of seven monoclonal antibody drugs (mAb 1-7) in different animal sera (mouse, rat and monkey) using TTPPVLDSDGSFFLYSK peptide as a quantitative surrogate peptide and TPEVTCVVVDVSHEDPEVK and VVSVLTVLHQDWLNGK as qualitative (confirmatory) peptides [13].
3.2 Immunocapture Reagent
Three types of capture reagents are typically used for hybrid LC-MS/MS assays: 1). Anti-idiotypic (anti-ID) antibody capture (specific), 2). Target antigen capture (specific), and 3). Protein A/G capture (non-specific). The choice of immunocapture reagent is often determined by availability of specific capture reagents and the sensitivity/selectivity requirements of the assay.
Specific capture reagents (Anti-ID antibodies or target antigens) are usually labeled (e.g. with Biotin) and immobilized to magnetic beads (e.g. Streptavidin magnetic beads). Protein A and protein G are bacterial immunoglobin (IgG) binding proteins. They are often immobilized on solid surfaces such as Sepharose™ or magnetic particles/beads. Protein A or G bind immunoglobulins from different species with different affinities and are used for IgG purification from different biomatrices [14]. Recombinant protein A/G can also be used to expand the binding activity [14].
Anti-Human IgG (Fc specific) antibodies, which are non-specific in human matrix but specific in preclinical matrices, have high selectivity for the Fc fragment of human IgG and often used for the analysis of human or humanized antibody-based therapeutics (mAbs, ADCs, Fc based fusion proteins/peptides) in pre-clinical species, with limited cross reactivity with endogenous animal IgG.
Protein A/G and biotinylated anti-human Fc/Streptavidin beads, which are commercially available, have been successfully used for quantifications of different human/humanized monoclonal antibody (mAb) biotherapeutics in non-human matrices. Protein A/G is considered non-selective and as such lower limits of quantitation are typically ≥ 0.050 µg/mL, while the use of an anti-human Fc immunocapture step often provides lower limits of quantitation ≤ 0.050 µg/mL.
Immunocapture using Protein A or G can also be extended to human matrix, which is advantageous when critical reagents, such as anti-ID antibodies, are not available. For example, a multiplex LBA-LC-MS/MS method for simultaneous quantification of seven therapeutic monoclonal antibodies (adalimumab, cetuximab, infliximab, rituximab, secukinumab, tocilizumab, and trastuzumab) in human plasma using protein G immunocapture has been reported [15]. Signature (proteotypic) surrogate peptides for each analyte were identified via in-silico analysis as described before [15] and used for quantification. The mAbs were either chimeric, humanized, or fully human IgG1 with different pharmacological targets. The LLOQs for the mAb therapeutics ranged from 1.0-5.0 µg/mL. The results not only showed the wide applicability of the generic protein G approach but also the multiplexing capability of using LC-MS/MS.
There are several important considerations that need to be considered and or evaluation regarding optimization of the immunocapture step. The amount of beads as well as bead type (Bead capacity), beads from different manufactures, ratio of biotinylated capture reagent to therapeutic and time profiling for capture should be studied and optimized during method development.
4.0 Generic Digestion Procedure
The standard digestion procedure includes: 1). denaturing with a surfactant such as Rapigest® or ProteaseMAX® to solubilize/unfold the protein, making the cleavage sites more accessible to digestion enzymes and improve the digestion efficiency (heat denaturation can be also applied with and without denaturing agent), 2). reduction using a reducing agent such as Tris(2-carboxyethyl) phosphine hydrochloride (TCEP) or Dithiothreitol (DTT) to reduce protein disulfide bonds (both denaturing and reduction can be carried out simultaneously), 3). alkylation of cysteine residues in proteins with Iodoacetic acid (IAA) or Iodoacetamide (IAM) to cap the reactive thiols, and 4). protein digestion using an appropriate enzyme such as trypsin.
4.1 Digestion Enzyme
Through the in-silico/ BLAST® analysis, it is possible to target a specific region of the protein by choosing the appropriate digestion enzyme (or a mixture of digestion enzymes) to generate a surrogate peptide that encompasses the region of interest. Trypsin is the most commonly used enzyme for protein quantitation due to its cleavage selectivity and reproducibility. Trypsin cleaves at the carboxyl terminal of lysine/arginine residues, accordingly, the tryptic peptides have basic residues at the C terminus, which is appropriate for reversed-phase liquid chromatography and MS detection [16].The recommended enzyme:protein ratio is 1:20 (w/w), and the most commonly used trypsin digestion protocol involves incubation at 37 °C in digestion buffers (pH ~ 8). The digestion time may range from as little as 1 hour to overnight [11,13,15]. Other digestion enzymes such as chymotrypsin, Asp-N®, Glu-C®, Lys-C®, protease K and pepsin have also been used in hybrid LC-MS/MS workflows. Lys-C/trypsin digestion has been applied to improve cleavage at lysine residues and Lys-C/Arg-C digestion has shown a better performance than trypsin and Lys-C/trypsin digestion [16].
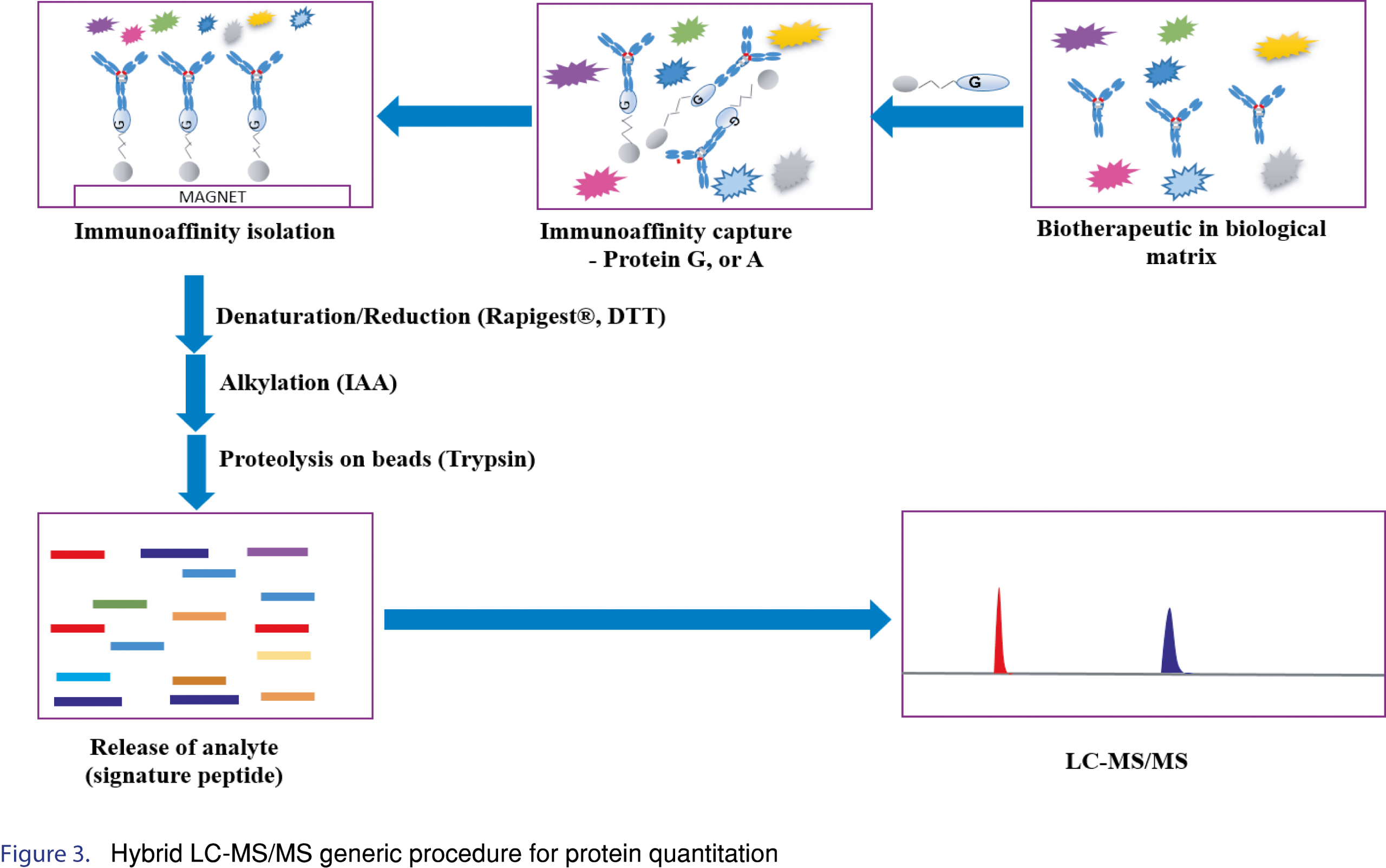
For protein quantitation by LC-MS, the speed and efficiency of the enzymatic digestion step is an important consideration for method development as well as for improving sample analysis throughput. The digestion efficiency is critical for the reproducibility and sensitivity of the assay. To ensure consistent digestion efficiency, denaturation, reduction and alkylation steps should be optimized in terms of reagent volume, time and temperature for each step. If the proteins are not appropriately denatured/reduced/alkylated, digestion efficiency may be negatively impacted, leading to low apparent digestion efficiencies or worse variable digestion efficiencies. Furthermore, after identification of an appropriate enzyme (as mentioned above), amount of enzyme (enzyme to therapeutic ratio), time and temperature for digestion should be assessed. Optimization of the digestion protocol to ensure consistent digestion efficiency is paramount since often times the internal standard is a stable isotopically labeled tryptic peptide (SIL-peptide), which is not digested. Internal standard considerations will be discussed further. An example of hybrid LC-MS/MS generic procedure for protein quantitation is shown in Figure 3.
5.0 Internal standard
A good internal standard can compensate for variabilities during sample preparation and digestion.
Four types of internal standards can be used in hybrid LC-MS/MS assays [17]:
- Stable-isotope labeled (SIL) therapeutic protein
- Analog Stable-isotope labeled (SIL) protein, such as SiluMAB™
- Extended or winged isotopically labeled surrogate peptide
- Isotopically labeled surrogate peptide
SIL-proteins are added before the capture step and have the advantage of being able to compensate for any variabilities associated with immunocapture, digestion, and detection. Although SIL-proteins are considered ideal internal standards, they are typically not commercially available, are expensive and timely to generate, and would be needed for each biotherapeutic [18]. Analog Stable-isotope labeled (SIL) proteins, such as SiluMAB™, while not identical to SIL-therapeutic proteins, can be used in non-selective immunocapture (Protein A or G, anti-human Fc) procedures. SiluMAB™ is produced by uniform incorporation of [13C6,15N4]-Arginine and [13C6,15N2]-Lysine amino acids and for several human IgG isotypes [12,13]. SiluMAB™ is added before the capture step and has the advantage of being able to compensate for any variabilities associated with immunocapture, digestion, and detection. An additional advantage is it is inexpensive, and readily commercially available. In addition SiluMAB™ could be considered a universal internal standard for human mAb, ADCs, or fusion protein therapeutics when developing bioanalytical method for pre-clinical species. Labeled extended surrogate peptides contain additional amino acid residues, typically 4-6 residues, on one or both ends of the peptide (N- and/or C-terminus) which can be enzymatically digested to produce the labeled surrogate peptide. They are relatively inexpensive and can be synthesized typically within three weeks. SIL-extended peptides are added after the immunocapture step and potentially help to reduce enzymatic digestion variability and LC/MS detection. Given the digestion efficiency between the intact protein and the extended peptide could be different, it is important to evaluate the consistency of the digestion efficiency, during method development, for both forms. Stable isotope-labeled peptide internal standards (SIL-IS) are typically added at the end of extraction (after digestion). They compensate for volume differences and matrix suppression/enhancement effects associated with LC-MS/MS detection. SIL-IS are very common for protein quantification and have the same advantages as the SIL-extended peptides in terms of costs and time to synthesize. Both SIL-extended peptides and SIL-peptides offer the scientist the ability to pick the number and location of the amino acids to be labeled. This allows flexibility to control the mass shift between the labeled and unlabeled peptide and to control having the labeled amino acid in the fragment ion (product ion).
6.0 Equipment
6.1 Beads Processor
Magnetic and Sepharose™ beads, coated with immobilized streptavidin and Protein A or G, are the most commonly used format in hybrid LC-MS/MS assays. Immunocapture using magnetic beads allows the target protein to be separated/enriched from matrix components by using the power of a magnet [14]. Beads are available from many different manufacturers, bead size, binding capacity, as well as surface modifications to minimize non-specific binding of unwanted proteins. Due to the available choices and the differences between beads, it is recommended that several types be evaluated, such as from different manufacturers, for selectivity, sensitivity and consistency.
There are two types of equipment that are primarily used for magnetic bead immunocapture enrichment/washing, microplate washers fitted with a flat magnet and magnetic particle processors. In essence, traditional plate washers remove the liquid (most but not all) from the beads. However, magnetic particle processors, such as the Thermo Scientific™ KingFisher™ Flex, use individual magnet heads to separate, wash and transfer the beads, i.e. beads removed from the liquid. While on the surface the difference may appear to be subtle, the use of the KingFisher™ can often help reduce variability associated with elution and/or digestion compared to a plate washer. Since plate washers leave residual and variable volumes of wash buffer “behind”, the residual buffer can impact the elution and digestion efficiency thus leading to increased variability.
Figures and Tables
[Click to enlarge]
6.2 Detection Instrument
LC-MS/MS based methods are a popular choice for the quantitation of biotherapeutics using a bottom up approach, which is based on proteolytic digestion of target biotherapeutics and using surrogate peptides for quantification. Due to their sensitivity, selectivity, and widespread use in laboratories, triple quadrupole mass spectrometers are the workhorse systems for bottom up quantitation. Although not as wide spread, high-resolution mass spectrometer instruments (HRMS) can also be used for both quantitation and characterization. In addition, HRMS offers enhanced mass selectivity that is not achievable using triple quadrupole systems, such as to accurately discriminate between an amidated and de-amidated peptide sequence.
In some cases, middle down and middle up approaches using selective digestion/agents (such as IdeZ®) to obtain protein subunits (e.g. Fab, Fc) can be applied. A middle-up LC–MS approach has been used for characterization of thiol-conjugated maleimidocaproyl-monomethyl auristatin F (mcMMAF) and valine-citrulline-monomethyl auristatin E (vcMMAE) antibody–drug conjugates (ADCs) and to calculate the drug–antibody ratio [18]. Biotherapeutics can also be directly analyzed by LC–MS using an intact level (Top-down) approach. Both middle down/up and top down approaches can offer additional structural information. However, due to the size, a high-resolution mass spectrometer should be used (e.g., qToF or Orbitrap instrument). While additional structural information can be provided by these techniques, it is often at the sacrifice of sensitivity. While the use of HRMS instrumentation has increased over the past few years, it is not as widespread as triple quadrupole instruments in terms of biotherapeutic quantitation.
6.3 Multiplex LC-MS/MS Assays for Biotherapeutics and related challenges
The number of protein-based therapeutics being developed are significantly increasing. From a bioanalytical perspective, the structural complexities of the therapeutics are becoming more challenging. Examples include ADCs with cleavable and non-cleavable linkers, bispecific antibodies, multi-domain fusion proteins, and emerging modalities such as Probody™ therapeutics. These molecules have additional challenges that extends beyond the “normal” challenges of assay development. For example, multi-domain fusion peptides may be needed to be able to accurately quantitate the concentrations based on each domain to ensure the molecule is intact and/or determine if the molecule has been catabolized to the various domains, which ultimately can help with explaining efficacy. The number of Oncology therapeutics is rapidly growing, and many treatments involve co-dosing biotherapeutics. In many cases the co-dosed therapeutics target the same antigen and have high degree of homology. As such, traditional LBA assays can suffer from cross-reactivity between the co-dosed therapeutics. By implementing a hybrid LC-MS/MS approach, reagent cross-reactivity is not an issue due to the resolving power of the mass spectrometer. Even with therapeutics that have >99% sequence homology, it is possible to accurately quantitate each therapeutic in a single assay by identifying surrogate peptides containing non-homologous sequences (even a single amino acid substitution could be enough to ensure appropriate mass selectivity).
Antibody drug conjugates are another example illustrating molecule complexity. Typically three PK assays are needed to assess safety and efficacy: 1). Total antibody, including conjugated antibody, partially and fully de-conjugated antibody, 2). total ADC (antibody-conjugated payload concentration (DAR sensitive) or conjugated-antibody concentration (not DAR sensitive)), and 3). free drug (unconjugated toxin) [19]. Depending on the ADC properties, it may be possible to measure the total antibody and total ADC concentrations in a single assay using hybrid LBA-LC-MS/MS (20).
7.0 Applications
Hybrid LC-MS/MS has been successfully applied to a wide range of different biotherapeutics such as:
- Monoclonal antibodies (mAbs)
- Bispecifics (bsmAbs, DART®s, BiTE®s)
- Antibody-drug conjugates (ADCs), bispecific drug conjugates
- Carrier conjugates (albumin, PEG)
- Fusion proteins/peptides
- Peptide and protein therapeutics and biomarkers
- Messenger RNA therapeutics™
In conclusion, hybrid LC-MS/MS workflows can be applied to a wide range of biotherapeutics:
An immunoaffinity approach using Protein A or G magneic beads can be used to enrich Fc containing biotherapeutics from human and animal matrix. Proteolysis with an appropriate enzyme, following standard protein denaturation, reduction, and alkylation processing steps. One or two signature/universal peptides are usually identified and used as surrogates for target protein quantitation. Different digestion enzymes can be used either alone or in combination to significantly increase the proverbial “development toolbox”.
- Anti-Human Fc antibodies and magnetic beads can be applied for selective isolation of Fc containing therapeutics from animal matrices (Pre-clinical studies). Followed by same procedure explained above.
- Immunoaffinity isolation by using biotinylated anti-ID antibody or target antigen and streptavidin beads can be applied to both human and animal matrices. The captured biotherapeutic will be denatured, reduced and digested following the same procedure above.
The complexities associated with biotherapeutics demands a clear understanding of what needs to be measured followed by strategies on how to measure. The use of hybrid LC-MS/MS expands the development strategy toolbox to enable such measurements.
7.0 Quality assurance in metabolomics using VAMS
8.0 Future outlook
8.0 References
1. M.N. Khan, J.W. Findlay (Eds), Ligand-binding assays development, validation, and implementation in the drug development arena, (Eds.), John Wiley & Sons. Hoboken, New Jersey, USA pages 2. (2009).
2. Furlong MT, Ouyang Z, Wu S, Tamura J, Olah T, Tymiak A, Jemal M. A universal surrogate peptide to enable LC-MS/MS bioanalysis of a diversity of human monoclonal antibody and human Fc-fusion protein drug candidates in pre-clinical animal studies. Biomed Chromatogr 26, 1024–1032 (2012). [CrossRef]
3. Dawes ML, Gu H, Wang J, Schuster AE, Haulenbeek J. Development of a validated liquid chromatography tandem mass spectrometry assay for a PEGylated adnectin in cynomolgus monkey plasma using protein precipitation and trypsin digestion. J Chromatogr B Anal Technol Biomed Life Sci 934, 1–7 (2013). [CrossRef]
4. Jiang H, Zeng J, Titsch C, Voronin K, Akinsanya B, Luo L, Shen H, Desai DD, Allentoff A, Aubry AF, Desilva BS, Arnold ME., Fully validated LC-MS/MS assay for the simultaneous quantitation of co-administered therapeutic antibodies in cynomolgus monkey serum. Anal Chem 85(20), 9859–67 (2013). [CrossRef]
5. Wang Y, Heilig JS. Differentiation and quantification of endogenous and recombinant-methionyl human leptin in clinical plasma samples by immunocapture/mass spectrometry. J Pharm Biomed Anal 70, 440-6 (2012). [CrossRef]
6. Xu K., Liu L, Maia M, Li J, Lowe J, Song A, Kaur SA, multiplexed hybrid LC-MS/MS pharmacokinetic assay to measure two co-administered monoclonal antibodies in a clinical study. Bioanalysis. 6(13), 1781–94 (2014). [CrossRef]
7. Hutchens TW, Yip T, New Desorption Strategies for the Mass Spectrometric Analysis of Macromolecules. Rapid Commun Mass Spec 7, 576-580 (1993). [CrossRef]
8. Nelson RW, Krone JR, Bieber AL, Williams P, Mass Spectrometric Immunoassay. Anal Chem 67, 1153-1158 (1995). [CrossRef]
9. Ewles M, Goodwin L, Bioanalytical approaches to analyzing peptides and proteins by LC–MS/MS. Bioanalysis 3(12), 1379–1397 (2011). [CrossRef]
10. Budhraja RH, Shah MA, Suthar M, Yadav A, Shah SP, Kale P, Asvadi P, Arasu MV, Al-Dhabi NA, Park CG, Kim Y, Kim HJ, Agrawal YK, Ravi. K. Krovidi RK, LC-MS/MS Validation Analysis of Trastuzumab Using dSIL Approach for Evaluating Pharmacokinetics. Molecules 21, 1464-1473 (2016). [CrossRef]
11. Cameron F, Whiteside G, Perry C. Ipilimumab. Drugs 71 (8), 1093-1104 (2011). [CrossRef]
12. Furlong MT, Zhao S, Mylott WR, Jenkins R, Gao M, Hegde V, Tamura J, Tymiak A,Jema M, Dual universal peptide approach to bioanalysis of human monoclonal antibody protein drug candidates in animal studies. Bioanalysis 5(11), 1363–1376 (2013). [CrossRef]
13. Kaur S, Liu L, Cortes DF, Shao J, Jenkins R, Mylott WR, Xu K, Validation of a biotherapeutic immunoaffinity-LC–MS/MS assay in monkey serum: ‘plug-and-play’ across seven molecules. Bioanalysis 8(15), 1565–1577 (2016). [CrossRef]
14. Safarik I, Safarikova M, Magnetic techniques for the isolation and purification of proteins and peptides. BioMagn Res Technol 2-7 (2004).
15. Willeman T, Jourdil J, Gautier-Veyret E, Bonaz B, Stanke-Labesque F, A multiplex liquid chromatography tandem mass spectrometry method for the quantification of seven therapeutic monoclonal antibodies: Application for adalimumab therapeutic drug monitoring in patients with Crohn’s disease. Anal Chim Acta 1067, 63-70 (2019). [CrossRef]
16. Wu Z, Huang J, Huang J, Li Q, Zhang X, Lys-C/Arg-C, a More Specific and Efficient Digestion Approach for Proteomics Studies. Anal Chem 90, 9700-9707 (2018). [CrossRef]
17. Ismaiel OA, Mylott Jr WR, Jenkins RG. Do we have a mature LC–MS/MS methodology for therapeutic monoclonal antibody bioanalysis? Bioanalysis 9(18), 1289–1292 (2017). [CrossRef]
18. Firth D, Bell L, Squires M, Estdale S, A rapid approach for characterization of thiol conjugated antibody–drug conjugates and calculation of drug–antibody ratio by liquid chromatography mass spectrometry, liquid chromatography. Anal Biochem 485, 34–42 (2015). [CrossRef]
19. Stevenson L, Garofolo F, Binodh DeSilva B, Isabelle, Dumont I, et. al. White Paper on recent issues in bioanalysis: ‘hybrid’ – the best of LBA and LCMS. Bioanalysis 5(23), 2903–2918 (2013) . [CrossRef]
20. Faria M, Peay M, Lam B et al. Rosenbaum. Multiplex LC-MS/MS Assays for Clinical Bioanalysis of MEDI4276, An Antibody-Drug Conjugate of Tubulysin Analogue Attached via Cleavable Linker to A Biparatopic Humanized Antibody against HER-2. Antibodies 8 (1), 11 (2019). [CrossRef]
All site content, except where otherwise noted, is licensed under a Creative Commons Attribution 4.0 License

